Basis-Update and Galerkin (BUG) rank-adaptive integration of a tree tensor network state¶
This notebook performs a Basis-Update and Galerkin (BUG) rank-adaptive integration of a tree tensor network state. The implementation in chemtensor is based on the publication:
Gianluca Ceruti, Christian Lubich, Dominik Sulz
Rank-adaptive time integration of tree tensor networks
[1]:
import numpy as np
from scipy.linalg import expm
import matplotlib.pyplot as plt
import chemtensor
[2]:
# maximum number of OpenMP threads (0 indicates that OpenMP is not available)
chemtensor.get_max_openmp_threads()
[2]:
16
Construct a Hamiltonian as TTNO from a list of operator chains¶
[3]:
# number of physical lattice sites
nsites_physical = 6
[4]:
# physical quantum numbers at each site
qsite = [1, 0, -1]
[5]:
# local operators
sp = np.array([[0., 1., 0.], [0., 0., 1.], [0., 0., 0.]])
sm = np.array([[0., 0., 0.], [1., 0., 0.], [0., 1., 0.]])
sz = np.diag([ 1., 0., -1.])
nb = np.diag([ 1., 0., 1.])
# operator map; the operator identifiers (OIDs) are the indices for this lookup-table
opmap = [np.identity(3), sp, sm, sz, nb]
[6]:
def crandn(size=None, rng: np.random.Generator=None):
"""
Draw random samples from the standard complex normal (Gaussian) distribution.
"""
if rng is None:
rng = np.random.default_rng()
# 1/sqrt(2) is a normalization factor
return (rng.normal(size=size) + 1j*rng.normal(size=size)) / np.sqrt(2)
[7]:
rng = np.random.default_rng(42)
# random kinetic coefficients
tkin = crandn(size=nsites_physical-1, rng=rng)
# random interaction coefficients
vint = list(rng.standard_normal(5))
# coefficient map; first two entries must always be 0 and 1;
# the coefficient identifiers (CIDs) are the indices for this lookup-table
coeffmap = [0, 1] + [c if orig else c.conj() for c in tkin for orig in [True, False]] + vint
[8]:
# operator chains, containing operator identifiers (OIDs) and coefficient identifiers (CIDs)
# referencing 'opmap' and 'coeffmap', respectively
chains = (
[chemtensor.OpChain([1, 2], [0, 1, 0], 2 + 2*i, istart=i) for i in range(nsites_physical - 1)] +
[chemtensor.OpChain([2, 1], [0, -1, 0], 2 + 2*i + 1, istart=i) for i in range(nsites_physical - 1)] +
[chemtensor.OpChain(rng.choice([0, 3, 4], size=4), [0, 0, 0, 0, 0],
2 + 2*len(tkin) + i,
istart=rng.integers(0, nsites_physical - 3)) for i in range(len(vint))])
In general, the integer quantum numbers are interleaved with the local operators to implement abelian symmetries (like particle number conservation). In practice, the quantum numbers endow the TTNO tensors with a sparsity pattern, handled internally by chemtensor.
To construct the TTNO, we need to define its tree topology.
[9]:
# tree topology:
#
# ╭──── 7 ────╮
# ╱ │ ╲
# ╱ │ ╲
# 0 3 6
# ╱ ╲ ╱ ╲
# ╱ ╲ ╱ ╲
# 1 2 4 5
#
tree_neighbors = [
[7], # neighbors of site 0
[3], # neighbors of site 1
[3], # neighbors of site 2
[1, 2, 7], # neighbors of site 3
[6], # neighbors of site 4
[6], # neighbors of site 5
[4, 5, 7], # neighbors of site 6
[0, 3, 6], # neighbors of site 7
]
Sites 0, …, 5 are physical sites, and the remaining sites 6, 7 are branching sites (with dummy physical legs of dimension 1).
[10]:
hamiltonian = chemtensor.construct_ttno_from_opchains("double complex", nsites_physical, tree_neighbors,
chains, opmap, coeffmap, qsite)
[11]:
# number of physical sites
hamiltonian.nsites_physical
[11]:
6
[12]:
# number of branching sites (in this example, sites 6 and 7)
hamiltonian.nsites_branching
[12]:
2
[13]:
# show virtual bond dimension between sites 3 and 7
hamiltonian.bond_dim(3, 7)
[13]:
9
[14]:
# show virtual bond dimension between sites 5 and 6
hamiltonian.bond_dim(5, 6)
[14]:
5
[15]:
# overall matrix representation of the TTNO
hamiltonian_mat = hamiltonian.to_matrix()
hamiltonian_mat.shape
[15]:
(729, 729)
[16]:
# check Hermitian symmetry
np.linalg.norm(hamiltonian_mat.conj().T - hamiltonian_mat)
[16]:
0.0
Construct a random initial TTNS¶
[17]:
# quantum numbers for all sites (physical and branching)
qsites = hamiltonian.nsites_physical * (qsite,) + hamiltonian.nsites_branching * ([0],)
[18]:
# logical overall quantum number sector of the state
qnum_sector = 2
[19]:
state = chemtensor.construct_random_ttns("double complex", nsites_physical, tree_neighbors,
qsites, qnum_sector, max_vdim=80, rng_seed=42, normalize=True)
[20]:
# maximum virtual bond dimension
state.max_bond_dim
[20]:
27
[21]:
# show virtual bond dimension between sites 3 and 7
state.bond_dim(3, 7)
[21]:
27
[22]:
# show virtual bond dimension between sites 0 and 7
state.bond_dim(0, 7)
[22]:
3
[23]:
# initial state as vector
state_init_vec = state.to_statevector()
[24]:
# should be normalized
np.linalg.norm(state_init_vec)
[24]:
0.9999999999999999
[25]:
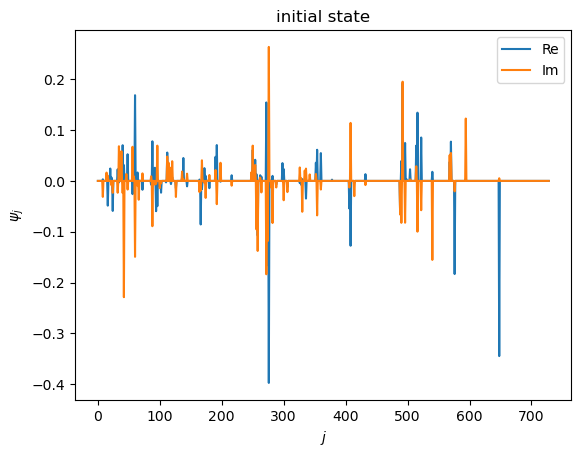
# visualize initial state
plt.plot(state_init_vec.real, label="Re")
plt.plot(state_init_vec.imag, label="Im")
plt.xlabel(r"$j$")
plt.ylabel(r"$\psi_j$")
plt.title("initial state")
plt.legend()
plt.show()
Perform BUG time integration¶
The BUG integrator updates the state in-place.
[26]:
# integration prefactor
prefactor = -0.1 - 0.8j
# time step
dt = 0.1
# number of time steps
nsteps = 5
# designated root node
i_root = 7
for i in range(nsteps):
chemtensor.bug_tree_time_step(state, hamiltonian, i_root, prefactor, dt, rel_tol_compress=1e-2)
[27]:
# quantum number sector remains unchanged
state.quantum_number_sector
[27]:
2
[28]:
# maximum bond dimension after time integration
state.max_bond_dim
[28]:
14
[29]:
# state vector after integration
state_vec = state.to_statevector()
[30]:
# state is not normalized due to complex integration prefactor
np.linalg.norm(state_vec)
[30]:
0.9851658533730621
[31]:
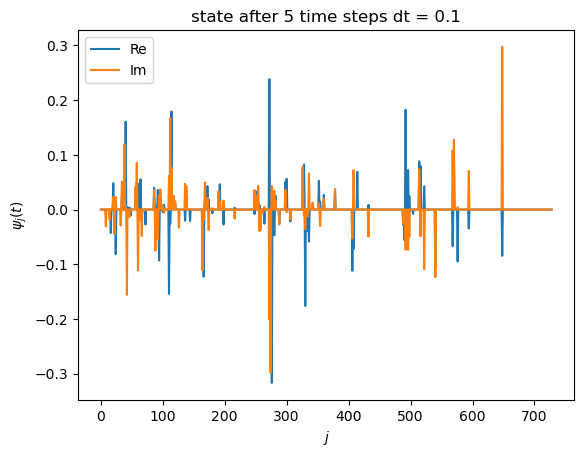
# visualize time-evolved state
plt.plot(state_vec.real, label="Re")
plt.plot(state_vec.imag, label="Im")
plt.xlabel(r"$j$")
plt.ylabel(r"$\psi_j(t)$")
plt.title(f"state after {nsteps} time steps dt = {dt}")
plt.legend()
plt.show()
[32]:
# reference solution
tmax = nsteps*dt # overall simulation time
state_ref = expm(prefactor * tmax * hamiltonian_mat) @ state_init_vec
[33]:
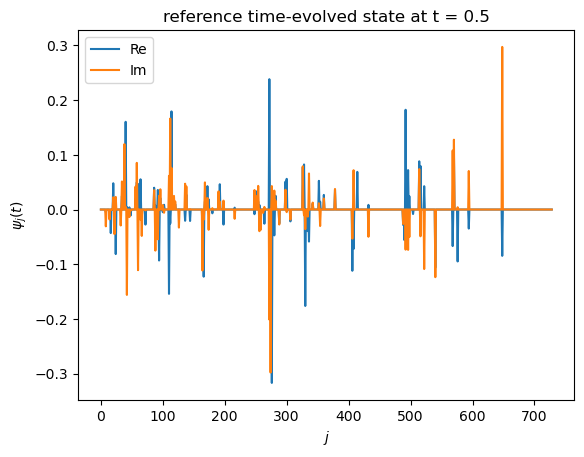
# visualize reference solution
plt.plot(state_ref.real, label="Re")
plt.plot(state_ref.imag, label="Im")
plt.xlabel(r"$j$")
plt.ylabel(r"$\psi_j(t)$")
plt.title(f"reference time-evolved state at t = {tmax}")
plt.legend()
plt.show()
[34]:
print("BUG integration error:", np.linalg.norm(state_vec - state_ref))
BUG integration error: 9.827381973754304e-05